Submitting Sequencing/Omics Data
Table of Contents
- Introduction
- 1. Join the Correct Group
- 2. Organize Your Data
- 3. Create Specimen Records
- 4. Create Metadata Records
- 5. Upload Files
- 6. Export for GEO Submission
- 7. Review and Submit
- Frequently Asked Questions
Introduction
This guide offers a clear pathway for submitting sequencing data to the ATLAS-D2K Data Explorer. For any questions, please contact help@atlas-d2k.org. Always ensure you fill out all relevant fields to make your data discoverable and reproducible.
These are some other training materials and other documentation you may find useful:
1. Join the Correct Group
- Follow these instructions.
- If unsure about the group to join, contact help@atlas-d2k.org.
- Remember to log in before submitting data.
2. Organize Your Data
Our system structures metadata records for efficient data discovery. They are organized as:
- Study: The top layer, describes high-level objectives and overall design of the experiments. It may contain one or several Experiments. Study-level analysis files can be uploaded to the Analysis Files section.
- Experiment: Describes the protocols, procedures, and experiment settings done to a set of Replicates. Linked records could include antibodies, custom metadata, and settings.
- Replicate: Provides bio-sample details and both biological and technical replicate numbers. Replicates include:
- All of the replicate-specific experimental assays, such as sequencing and analysis files, are uploaded under the File section.
- A link to a Specimen record.
- For single cell RNA-Seq, you may also add a Single Cell Metrics record summarizing the statistics of a Replicate.
- Replicate: Provides bio-sample details and both biological and technical replicate numbers. Replicates include:
- Experiment: Describes the protocols, procedures, and experiment settings done to a set of Replicates. Linked records could include antibodies, custom metadata, and settings.
2.1 Metadata Model for Sequencing Data
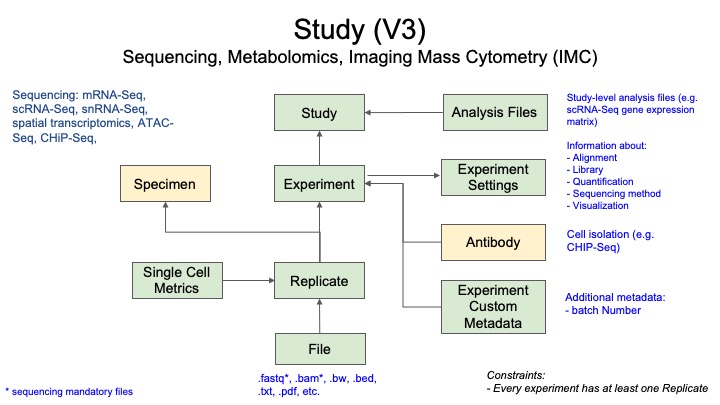
- Green boxes are the records directly related to this assay type.
- Yellow boxes indicate links to other assay types (in this case Specimen and Antibody, which have their own metadata requirements).
3. Create Specimen Records
For each tissue slide used, connect it to a Specimen record. Create these now and keep RID numbers handy for linking later. Comprehensive specimen submission guidelines are available here.
To describe the relationships between slides and sections of slides, use the Parent Specimen field as described in the documentation.
4. Create Metadata Records
4.1. Create Study Records
- Use this link to log in and directly create a Study record: https://www.atlas-d2k.org/chaise/recordedit/#2/RNASeq:Study
- You can also go to any Study page and click
Createin the record header (you must be logged in to see theCreatebutton).
- You can also go to any Study page and click
- Complete as many fields as are appropriate for your data and save.
- On the new Study record, scroll to the Study Analysis File section and click
Addto add analysis files associated with your study.
4.2. Create Experiment Records
- On the Study record, navigate to the Experiments section and click
Add Record. - Fill in the fields describing your experiment and save.
- On the newly created Experiment, scroll down and link additional records (for example, use Experiment Antibodies to link to the antibodies used for cell isolation in the experiment), and Experiment Settings to enter the details related to processing the data.
4.3. Create Replicate Records
- From the Experiment record, scroll down to the Replicate section and click
Add record. - Link to one of the Specimen records you created earlier. You can search the RIDs you recorded earlier.
- Add the Biological and Technical Replicate Numbers:
- The first replicate of an experiment will use the number
1for both Biological and Technical Replicate numbers.
- The first replicate of an experiment will use the number
- Fill in all other fields relevant to your data.
- After completion, save the record.
- On the newly created Replicate record, link additional records:
- Files: If you are only adding a few data files, scroll down to the Files table and click
Add. To upload multiple files at the same time, you can click the plus sign (+) to add more forms. If you are uploading many or very large files, we recommend using the bulk upload method described below. - Single Cell Metrics: For single cell RNASeq data, scroll down to the Single Cell Metrics table and click
Add recordto include this information.
- Files: If you are only adding a few data files, scroll down to the Files table and click
4.4. Record Status and Curation Status Fields
Here is helpful information about the Record Status and Curation Status fields:
- Incomplete records will indicate missing required information. The Record Status field will list which information is missing.
- The default value for the Curation Status field is “In preparation” and is not publicly available.
- Once you are finished with your data, change the status to “Submitted” for biocurator review (it’s a good idea to send the ATLAS-D2K Center a quick email to let you know it’s ready for review). Upon approval, we will change the status to “Release”, at which point the data is now publicly available.
- All records (e.g. Experiments, Replicates, Specimen, Files) associated with study inherit the same Curation Status as their Study. Once the Study is released, all the related records will also be released.
5. Upload Files
The .fastq and .bam files are mandatory. However, data submitters are encouraged to also upload corresponding analysis files if they are available.
5.1. Review Supported File Extensions
Check the provided table for approved extensions and descriptions.
| Extension | File Type | Description (will appear in file caption) | mandatory |
|---|---|---|---|
.R1.fastq.gz |
FastQ | F reads | mandatory |
.R2.fastq.gz |
FastQ | R reads | mandatory for Paired-End |
.I1.fastq.gz |
FastQ | I1 reads | optional |
.bam |
bam | alignment | mandatory |
.bed |
bed | positive regions | optional |
.bw |
bigWig | visualization track | optional |
.tpm.csv, .tmp.tsv, or .tpm.txt |
csv, tsv, or txt respectively | expression value | optional |
.rpkm.csv, .rpkm.tsv, or .rpkm.txt |
csv, tsv, or txt respectively | expression value | optional |
.fpkm.csv, .fpkm.tsv, or .fpkm.txt |
csv, tsv, or txt respectively | expression value | optional |
.ReadsPerGene.csv, .ReadsPerGene.tsv, or .ReadsPerGene.txt |
csv, tsv, or txt respectively | Reads per gene | optional |
5.2. Browser Upload Method
This method is suitable for fewer replicates and smaller files. For a quick guide:
- Study record: Add analysis files using the Study Analysis File section.
- Replicate record: Add sequencing and analysis files to the appropriate Replicate record via the File section.
- Normally, users will need to upload the actual data files to the Hub. For sequencing files that are archived in other permanent repositories (e.g. GEO, dbGaP), you also have the option to enter a URL for to the archive.
- For human-protected sequencing file stored in dbGaP, please provide a
dbGaP Accession ID.
5.3. Bulk Upload with Client Tools
Use this method for many replicates or larger files (more than 10 files or files are larger than 5GB).
You will need to:
- Install the client tools on your system. There are pre-packaged installers for MacOS and Windows available as well as an installer via pip for Linux desktops or remote servers.
- Organize your directory structure and use a naming convention for your files (You will need to note the Internal IDs you used in your Studies and Experiments).
- Run the tool for your files to set up a submission job for the files.
This is a very secure and stable service, and if the job is interrupted, the program will be able to retry submission until success. If the job still fails, you can re-run the program and it will be able to tell which files were already successfully uploaded and skip them.
If your directory structure and naming convention are correct, the files will be automatically attached to the correct Replicate records.
For full instructions, go to Bulk Uploading Files with DERIVA Client Tools.
6. Export for GEO submission
Once you’ve created your sequencing study and uploaded your data, you can then export to a file for submission to GEO. See the Exporting to submit to GEO section of the Exporting Data page.
7. Review and Submit
7.1. Data Submission Dashboard
The dashboard gives an overview of your submitted records. Ensure everything’s complete and accurate.
The monthly data submission dashboard is available on the ATLAS-D2K data browser.
Before public release, the curation team reviews submissions. Once reviewed, the status becomes “Release” and data’s accessible.
Frequently Asked Questions
Question: 10X generates fastq files in the form of *.R1_001.fastq.gz (e.g. my_single_cell.R1_001.fastq.gz). How do I rename file names in bulk from *.R1_001.fastq.gz to *.R1.fastq.gz?
Answer: You can run the following command on the shell prompt to rename files in bulk.
# to change *.R1_001.fastq.gz to *.R1.fastq.gz
> for file in *.fastq.gz; do mv -v "$file" $(echo "$file" | sed 's/.R1_001.fastq.gz$/.R1.fastq.gz/'); done
Question: CellRange software expects the sequencing files to be in the form of *.R1_001.fastq.gz. How do I change the file names from *.R1.fastq.gz to *.R1_001.fastq.gz?
Answer: You can run the following command on the shell prompt to rename files in bulk.
# to change *.R1.fastq.gz to *.R1_001.fastq.gz
> for file in *.fastq.gz; do mv -v "$file" $(echo "$file" | sed 's/.R1.fastq.gz$/.R1_001.fastq.gz/'); done
For any queries, contact help@atlas-d2k.org. Our team’s always ready to support your data submission needs.